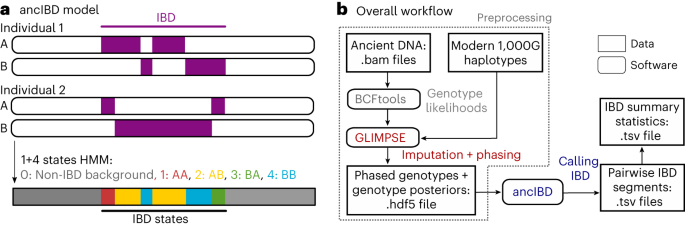
Found a GLIMPSE VCF, after filtering I extracted, edited the heads, and tested the Antonio_2019 samples, ... we know that R53 and R54 are related.
Some Resuts:
IBD
iid1 | iid2 | max_IBD | sum_IBD>8.0 | n_IBD>8.0 | sum_IBD>12.0 | n_IBD>12.0 | sum_IBD>16.0 | n_IBD>16.0 | sum_IBD>20.0 | n_IBD>20.0 |
R53 | R54 | 28.0063986778259 | 80.6819975376129 | 4 | 72.3859965801239 | 3 | 72.3859965801239 | 3 | 72.3859965801239 | 3 |
R54 | R64 | 33.1764996051788 | 75.3223031759262 | 3 | 64.697203040123 | 2 | 64.697203040123 | 2 | 64.697203040123 | 2 |
R52 | R53 | 32.5489997863769 | 62.9499077796936 | 3 | 53.7110030651093 | 2 | 53.7110030651093 | 2 | 53.7110030651093 | 2 |
R53 | R64 | 53.3635973930359 | 53.3635973930359 | 1 | 53.3635973930359 | 1 | 53.3635973930359 | 1 | 53.3635973930359 | 1 |
R9 | R17 | 12.1754944324493 | 21.9883859157562 | 2 | 12.1754944324493 | 1 | 0 | 0 | 0 | 0 |
R11 | R15 | 10.3309035301209 | 19.3359971046448 | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
R54 | R60 | 12.4449014663696 | 12.4449014663696 | 1 | 12.4449014663696 | 1 | 0 | 0 | 0 | 0 |
CH_ALL
Start | End | StartM | EndM | length | lengthM | ch | iid1 | iid2 |
4606 | 18543 | 0.577737 | 1.41346001625061 | 13937 | 0.835723042488098 | 15 | R53 | R54 |
17905 | 31857 | 1.186976 | 1.99431502819061 | 13952 | 0.807339072227478 | 1 | R53 | R54 |
17011 | 26303 | 1.140008 | 1.72522497177124 | 9292 | 0.585216999053955 | 1 | R54 | R64 |
17938 | 26303 | 1.188959 | 1.72522497177124 | 8365 | 0.536265969276428 | 1 | R53 | R64 |
10466 | 22910 | 0.627808 | 1.16144394874573 | 12444 | 0.533635973930359 | 3 | R53 | R64 |
717 | 8656 | 0.095289 | 0.584698975086212 | 7939 | 0.489409975707531 | 20 | R53 | R54 |
2607 | 8377 | 0.224026 | 0.659416019916534 | 5770 | 0.435390025377274 | 16 | R53 | R54 |
20 | 4130 | 0.014738 | 0.448329985141754 | 4110 | 0.433591985143721 | 17 | R53 | R64 |
12222 | 17141 | 0.789812028408051 | 1.17709004878998 | 4919 | 0.387278020381927 | 18 | R53 | R54 |
27088 | 33001 | 1.29929494857788 | 1.68000900745392 | 5913 | 0.380714058876038 | 8 | R53 | R54 |
5872 | 8528 | 0.649466 | 1.02555799484253 | 2656 | 0.37609201669693 | 19 | R54 | R64 |
4035 | 10914 | 0.308542 | 0.656117022037506 | 6879 | 0.347575008869171 | 7 | R53 | R54 |
836 | 6893 | 0.132061 | 0.478502005338669 | 6057 | 0.346441000699997 | 14 | R53 | R64 |
VCF files:

erda.ku.dk
some example:
... after filtering:
VCFs Head Editing:
bcftools head Romans_1.vcf.gz > 1header.hr
bcftools head Romans_2.vcf.gz > 2header.hr
bcftools head Romans_3.vcf.gz > 3header.hr
...and so on
Edit all Heads with the one below (copy and paste), replace
##contig=<ID=1> to 2header.hr with ##contig=<ID=
2> save it, ...and so on.
##fileformat=VCFv4.2
##FILTER=<ID=PASS,Description="All filters passed">
##fileDate=30/10/2021 - 05:10:05
##source=GLIMPSE_phase v1.0.0
##contig=<ID=
1>
##INFO=<ID=RAF,Number=A,Type=Float,Description="ALT allele frequency in the reference panel">
##INFO=<ID=AF,Number=A,Type=Float,Description="ALT allele frequency computed from DS/GP field across target samples">
##INFO=<ID=INFO,Number=A,Type=Float,Description="Imputation information or quality score">
##INFO=<ID=BUF,Number=A,Type=Integer,Description="Is it a variant site falling within buffer regions? (0=no/1=yes)">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Unphased genotypes">
##FORMAT=<ID=DS,Number=1,Type=Float,Description="Genotype dosage">
##FORMAT=<ID=GP,Number=3,Type=Float,Description="Genotype posteriors">
##FORMAT=<ID=HS,Number=1,Type=Integer,Description="Sampled haplotype pairs packed into intergers (max: 16 pairs, see NMAIN header line)">
##NMAIN=15
##INFO=<ID=pan_troglodytes,Number=1,Type=String,Description="allele observed in pan_troglodytes">
##FORMAT=<ID=PL,Number=G,Type=Integer,Description="List of Phred-scaled genotype likelihoods">
##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Allelic depths (high-quality bases)">
##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele count in genotypes">
##INFO=<ID=MASK_1000G,Number=0,Type=Flag,Description="SNP is in 1000G strict mask region">
##INFO=<ID=AN,Number=1,Type=Integer,Description="Total number of alleles in called genotypes">
##reference=ftp://ftp.1000genomes.ebi.ac.uk//vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz
##bcftools_viewVersion=1.13+htslib-1.13
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT R1 R2 R3 R4 R5 R6 R7 R8 R9 R10 R11 R15 R16 R17 R18 R19 R22 R24 R25 R26 R27 R28 R29 R30 R31 R32 R33 R34 R35 R36 R37 R38 R39 R40 R41 R42 R43 R44 R45 R47 R49 R50 R51 R52 R53 R54 R55 R56 R57 R58 R59 R60 R61 R62 R63 R64 R65 R66 R67 R68 R69 R70 R71 R72 R73 R75 R76 R78 R80 R81 R104 R105 R106 R107 R108 R109 R110 R111 R113 R114 R115 R116 R117 R118 R120 R121 R122 R123 R125 R126 R128 R130 R131 R132 R133 R134 R136 R137 R435 R436 R437 R473 R474 R475 R835 R836 R850 R851 R969 R970 R973 R1014 R1015 R1016 R1021 R1219 R1220 R1221 R1224 R1283 R1285 R1286 R1287 R1288 R1289 R1290 R1543 R1544 R1545 R1547 R1548 R1549 R1550 R1551
Now we replace the Heads:
bcftools reheader -h 1header.hr Romans_1.vcf.gz > Romans_chr1.vcf.gz
bcftools reheader -h 2header.hr Romans_2.vcf.gz > Romans_chr2.vcf.gz
bcftools reheader -h 3header.hr Romans_3.vcf.gz > Romans_chr3.vcf.gz
... and so on
")
 github.com
github.com