-
Don't want to see ads? Install an adblocker like uBlock Origin or use a Europe-based privacy-friendly browser like Vivaldi or Mullvad.
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.
You should upgrade or use an alternative browser.
Admixtools admixtools2 TUTORIAL for WINDOWS.
- Thread starter eupator
- Start date
qh777
Regular Member
- Messages
- 952
- Reaction score
- 572
- Points
- 93
- Ethnic group
- English, Scottish, Irish, French, German, Belgian
- Y-DNA haplogroup
- J2a1a1a2b2/J-FTC5373
- mtDNA haplogroup
- H/ H1/H1ao
** R
** data
*** moving datasets to lazyload DB
** inst
** byte-compile and prepare package for lazy loading
Note: break used in wrong context: no loop is visible
** help
*** installing help indices
*** copying figures
** building package indices
** installing vignettes
** testing if installed package can be loaded from temporary location
** testing if installed package can be loaded from final location
** testing if installed package keeps a record of temporary installation path
* DONE (admixtools)
Got it to work in the R Console. Within Rstudio itself I was getting errors
** data
*** moving datasets to lazyload DB
** inst
** byte-compile and prepare package for lazy loading
Note: break used in wrong context: no loop is visible
** help
*** installing help indices
*** copying figures
** building package indices
** installing vignettes
** testing if installed package can be loaded from temporary location
** testing if installed package can be loaded from final location
** testing if installed package keeps a record of temporary installation path
* DONE (admixtools)
Got it to work in the R Console. Within Rstudio itself I was getting errors
Celtion
Regular Member
- Messages
- 77
- Reaction score
- 60
- Points
- 18
For quite some months I've spent countless hours experimenting with G25 to see how well it can replicate admixture results from formal studies that utilise qpAdm and ADMIXTURE etc. I agree that G25 can be hit and miss. The problem is there is no way to validate the results except by having pre-existing knowledge.Check out this thread:
G25 - g25 VS qpadm/admixtools2 comparison.
Had some time to kill so here goes. These are my best ‘mixed mode’ results for each period for G25 Illustrative DNA. The raw dna file that was used to produce these results was an extract from my Whole Genome Sequence Nebula Genomics file, that has 99.9% SNP coverage with eurogenes’ template...www.eupedia.com
In addition to @eupator there's a few other members that could help you out.
There's times when it does replicate qpAdm, and times when it doesn't. Frankly, you should endeavor to use admixtools for optimal analysis. It is the suite of tools used by academics for peer-reviewed studies. Once you get it set up, it becomes second nature. I use AI to help me figure it out.
Anyway I've decided to learn ADMIXTURE for the time being and once I've got the hang of it I might move on to learn qpAdm. BTW I've noticed in this thread there's a few mentions of converting Eigenstrat format files to Plink format. Is there any benefit of this since qpAdm supports Eigenstrat files, doesn't it?
Jovialis
Advisor
- Messages
- 9,901
- Reaction score
- 6,809
- Points
- 113
- Ethnic group
- Italian
- Y-DNA haplogroup
- R1b-PF7566>Y227216
- mtDNA haplogroup
- H6a1b7
With eigenstrat you can take advantage of other tools, notably smartpca. Which is the same PCA that is used in many studies.For quite some months I've spent countless hours experimenting with G25 to see how well it can replicate admixture results from formal studies that utilise qpAdm and ADMIXTURE etc. I agree that G25 can be hit and miss. The problem is there is no way to validate the results except by having pre-existing knowledge.
Anyway I've decided to learn ADMIXTURE for the time being and once I've got the hang of it I might move on to learn qpAdm. BTW I've noticed in this thread there's a few mentions of converting Eigenstrat format files to Plink format. Is there any benefit of this since qpAdm supports Eigenstrat files, doesn't it?
However, I myself have tried converting back from PLINK to see where I plot. But I run into an issue where the file becomes massive for some reason. I haven't been able to figure it out, and I've asked around, but haven't found the answer. Like yourself, I am self-taught, and still learning.
Welcome to the forum btw!
Celtion
Regular Member
- Messages
- 77
- Reaction score
- 60
- Points
- 18
Thanks! Yeah I've downloaded the Reich dataset (the HO one) which of course is in Eigenstrat. Since ADMIXTURE also supports Eigenstrat format then I don't see much of a reason to convert the dataset to Plink. I just need to learn to create subsets of the Reich dataset. I notice however that the ADMIXTURE tutorials I've found tend to use Plink files – possibly because they were using an older version of ADMIXTURE which didn't support Eigenstrat, and/or because some of the operations in the tutorials would utilise Plink.With eigenstrat you can take advantage of other tools, notably smartpca. Which is the same PCA that is used in many studies.
However, I myself have tried converting back from PLINK to see where I plot. But I run into an issue where the file becomes massive for some reason. I haven't been able to figure it out, and I've asked around, but haven't found the answer. Like yourself, I am self-taught, and still learning.
Welcome to the forum btw!
Btw when you converted back from the Plink file, did you convert to Eigenstrat or VCF? (I read that VCF files are quite larger than the other formats).
There is easy way how to avoid this.With eigenstrat you can take advantage of other tools, notably smartpca. Which is the same PCA that is used in many studies.
However, I myself have tried converting back from PLINK to see where I plot. But I run into an issue where the file becomes massive for some reason. I haven't been able to figure it out, and I've asked around, but haven't found the answer. Like yourself, I am self-taught, and still learning.
Welcome to the forum btw!
You have to filter the samples being merged to the large dataset to only those SNPs already existing in the large dataset, else the extraneous SNPs in the sample that don't occur in the large forces the large to add No Calls to every sample in it swelling the dataset.
#
# This example would gather the SNPs in the primary and write them out. Then you use it to filter the sample, in this case my sample.
#
# You'd then merge this new filtered sample dataset with the primary like normal.
# plink --allow-no-sex --bfile v52.2_1240K_public --write-snplist --out v52.2_1240K_clean
# plink --23file PLg.txt --extract v52.2_1240K_clean.snplist --make-bed --out PLg_v54p1_genome
getwd()
# system("plink --allow-no-sex --bfile v52.2_1240K_public --write-snplist --out v52.2_1240K_clean ")
system("plink --bfile S2949 --extract v52.2_1240K_clean.snplist --make-bed --out S2949_filtered")
system("plink --bfile S2949_filtered --bmerge v52.2_1240K_public.bed v52.2_1240K_public.bim v52.2_1240K_public.fam --out H
 ut")
ut")--write-snplist - is the option to create clean snip list . Only the snips in this list will be used for the merge. This are the snips in the large dataset. Because the new file that you will merge may have some other snips or the names for the snips may be different which is also causing some issues. I noticed that some files may have different names for the snips, depending on the format.
Jovialis
Advisor
- Messages
- 9,901
- Reaction score
- 6,809
- Points
- 113
- Ethnic group
- Italian
- Y-DNA haplogroup
- R1b-PF7566>Y227216
- mtDNA haplogroup
- H6a1b7
Thanks! I did this when merging previously.There is easy way how to avoid this.
You have to filter the samples being merged to the large dataset to only those SNPs already existing in the large dataset, else the extraneous SNPs in the sample that don't occur in the large forces the large to add No Calls to every sample in it swelling the dataset.
#
# This example would gather the SNPs in the primary and write them out. Then you use it to filter the sample, in this case my sample.
#
# You'd then merge this new filtered sample dataset with the primary like normal.
# plink --allow-no-sex --bfile v52.2_1240K_public --write-snplist --out v52.2_1240K_clean
# plink --23file PLg.txt --extract v52.2_1240K_clean.snplist --make-bed --out PLg_v54p1_genome
getwd()
# system("plink --allow-no-sex --bfile v52.2_1240K_public --write-snplist --out v52.2_1240K_clean ")
system("plink --bfile S2949 --extract v52.2_1240K_clean.snplist --make-bed --out S2949_filtered")
system("plink --bfile S2949_filtered --bmerge v52.2_1240K_public.bed v52.2_1240K_public.bim v52.2_1240K_public.fam --out H
--write-snplist - is the option to create clean snip list . Only the snips in this list will be used for the merge. This are the snips in the large dataset. Because the new file that you will merge may have some other snips or the names for the snips may be different which is also causing some issues. I noticed that some files may have different names for the snips, depending on the format.
What I meant is after it is already in PLINK format the size is good, and comparable to the previous eigenstrat format; converting back to eigenstrat from PLINK it becomes like 10x bigger than the original eigenstrat file, despite only having 1 sample added (with no extra SNPs, just the ones native to the original eigenstrat.)
At any rate, thanks for that information, because it will be useful when i re-merge my sample when the new updates for AADR come out.
I think I have seen that also.Thanks! I did this when merging previously.
What I meant is after it is already in PLINK format the size is good, and comparable to the previous eigenstrat format; converting back to eigenstrat from PLINK it becomes like 10x bigger than the original eigenstrat file, despite only having 1 sample added (with no extra SNPs, just the ones native to the original eigenstrat.)
At any rate, thanks for that information, because it will be useful when i re-merge my sample when the new updates for AADR come out.
Celtion
Regular Member
- Messages
- 77
- Reaction score
- 60
- Points
- 18
For mobile you are limited to pca-based admixture calculators and oracles such as G25. To run anything like ADMIXTURE or qpAdm which are academic standard you need a Linux/Unix based environment on a desktop or laptop.What's the most accurate thing for mobile users? I use g25, but I'm reading its not reliable at all.
Celtion
Regular Member
- Messages
- 77
- Reaction score
- 60
- Points
- 18
Hi Jovialis,I currently have the 1240K+HO version which I converted to PLINK format.
If you want to add your own DNA, you first have to make sure the raw data is in 23andme format, and aligned to HG19 format. That is the format that AADR uses.
You then have to use PLINK to convert the AADR files from eigenstrat format to PLINK format. Then convert your 23andme raw data to PLINK format, and do a one sided merge in PLINK, making sure to only include SNPs native to AADR.
It is a bit of a complex process, I used Chatgpt 4.0 to help me out.
What do you mean by a one-sided merge in Plink? Also is there an issue with merging snps that aren't already in the AADR dataset?
So far I've avoided Plink for merging because it always fails and I'd prefer not to go through the convoluted process to deal with the strand-flipping. I merge the datasets in eigenstrat format using mergeit as it automatically sorts out those issues during the merge. I then convert back to Plink for QC filtering and LD pruning etc. But if there is a particular benefit in merging in Plink that is not mergeit I might try the Plink way, however complicated.
Last edited:
eupator
destroyer of delusions
- Messages
- 509
- Reaction score
- 287
- Points
- 63
- Ethnic group
- Rhōmaiōs (Rumelia + Anatolia)
This is is how you merge with PLINK:
My raw dna file in this example is eupator.txt in the 23ame format, if it's not in that format, convert it with dna kit studio or similar.
That will create the bed filed eupator.bed. Use an editor to rename your sample in the file to your liking.
Then use your converted .bed dataset to merge. In this example it's v54_HO_public.
The merged file is 54_HO_public_merged.
If you get strand inconsistency problems you can solve them, as they appear, with the following:
Your new merged dataset is v54_HO_public_final, you can use it straight away with at2 as at2 reads plink files.
My raw dna file in this example is eupator.txt in the 23ame format, if it's not in that format, convert it with dna kit studio or similar.
Code:
plink --23file eupator.txt --make-bed --out eupatorThat will create the bed filed eupator.bed. Use an editor to rename your sample in the file to your liking.
Then use your converted .bed dataset to merge. In this example it's v54_HO_public.
Code:
plink --allow-no-sex --bfile v54_HO_public --bmerge eupator --out v54_HO_public_eupatorThe merged file is 54_HO_public_merged.
If you get strand inconsistency problems you can solve them, as they appear, with the following:
Code:
plink --allow-no-sex --bfile eupator --flip v54_HO_public_eupator-merge.missnp --make-bed --out eupator_flipped
plink --allow-no-sex --bfile v54_HO_public --bmerge eupator_flipped --make-bed --out v54_HO_public_eupator2
plink --allow-no-sex --bfile eupator_flipped --exclude v54_HO_public_eupator2-merge.missnp --make-bed --out eupator_filtered
plink --allow-no-sex --bfile v54_HO_public --bmerge eupator_filtered --make-bed --out v54_HO_public_finalYour new merged dataset is v54_HO_public_final, you can use it straight away with at2 as at2 reads plink files.
Last edited:
Celtion
Regular Member
- Messages
- 77
- Reaction score
- 60
- Points
- 18
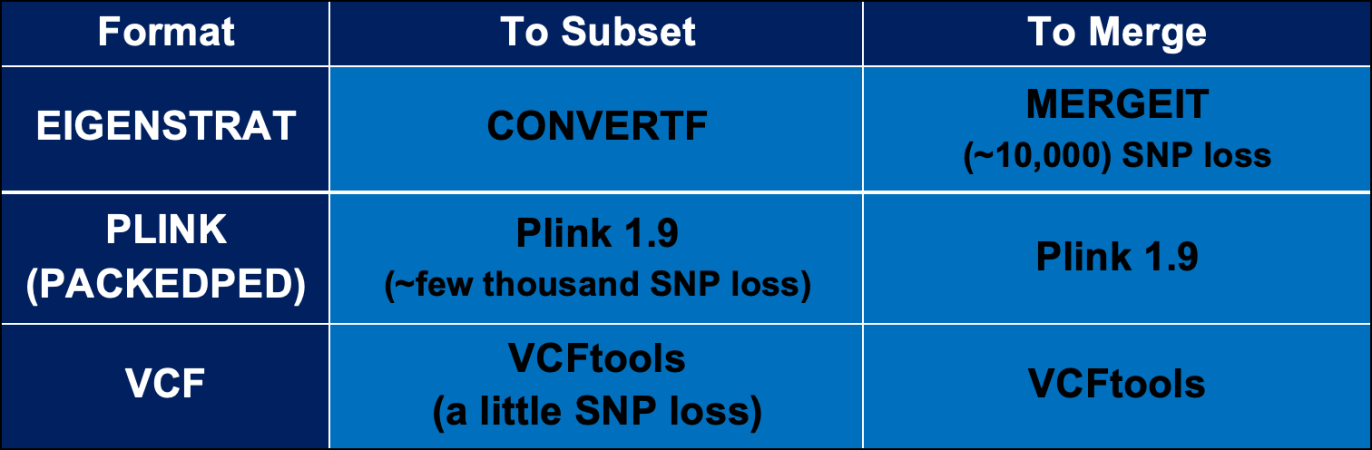
Thanks. I'd previously only seen the examples in the online PLINK 1.9 instructions. Yours was a bit different in that you did one flip and one exclude. I had been wondering what the benefit of merging in PLINK is when MERGEIT automatically checks and fixes strand incompatibilities during the merge without having to remedy the problem with more commands. Perhaps it's to do with what the chart below says. PLINK doesn't shed the amount of SNPs that MERGEIT does. I might experiment and compare.This is is how you merge with PLINK:
My raw dna file in this example is eupator.txt in the 23ame format, if it's not in that format, convert it with dna kit studio or similar.
Code:plink --23file eupator.txt --make-bed --out eupator
That will create the bed filed eupator.bed. Use an editor to rename your sample in the file to your liking.
Then use your converted .bed dataset to merge. In this example it's v54_HO_public.
Code:plink --allow-no-sex --bfile v54_HO_public --bmerge eupator --out v54_HO_public_eupator
The merged file is 54_HO_public_merged.
If you get strand inconsistency problems you can solve them, as they appear, with the following:
Code:plink --allow-no-sex --bfile eupator --flip v54_HO_public_eupator-merge.missnp --make-bed --out eupator_flipped plink --allow-no-sex --bfile v54_HO_public --bmerge eupator_flipped --make-bed --out v54_HO_public_eupator2 plink --allow-no-sex --bfile eupator_flipped --exclude v54_HO_public_eupator2-merge.missnp --make-bed --out eupator_filtered plink --allow-no-sex --bfile v54_HO_public --bmerge eupator_filtered --make-bed --out v54_HO_public_final
Your new merged dataset is v54_HO_public_final, you can use it straight away with at2 as at2 reads plink files.
Attachments
Celtion
Regular Member
- Messages
- 77
- Reaction score
- 60
- Points
- 18
BTW do you know how to pseudo-haploidize the data? (preferably in PLINK or EIGENSTRAT format)This is is how you merge with PLINK:
My raw dna file in this example is eupator.txt in the 23ame format, if it's not in that format, convert it with dna kit studio or similar.
Code:plink --23file eupator.txt --make-bed --out eupator
That will create the bed filed eupator.bed. Use an editor to rename your sample in the file to your liking.
Then use your converted .bed dataset to merge. In this example it's v54_HO_public.
Code:plink --allow-no-sex --bfile v54_HO_public --bmerge eupator --out v54_HO_public_eupator
The merged file is 54_HO_public_merged.
If you get strand inconsistency problems you can solve them, as they appear, with the following:
Code:plink --allow-no-sex --bfile eupator --flip v54_HO_public_eupator-merge.missnp --make-bed --out eupator_flipped plink --allow-no-sex --bfile v54_HO_public --bmerge eupator_flipped --make-bed --out v54_HO_public_eupator2 plink --allow-no-sex --bfile eupator_flipped --exclude v54_HO_public_eupator2-merge.missnp --make-bed --out eupator_filtered plink --allow-no-sex --bfile v54_HO_public --bmerge eupator_filtered --make-bed --out v54_HO_public_final
Your new merged dataset is v54_HO_public_final, you can use it straight away with at2 as at2 reads plink files.
eupator
destroyer of delusions
- Messages
- 509
- Reaction score
- 287
- Points
- 63
- Ethnic group
- Rhōmaiōs (Rumelia + Anatolia)
No, sorry.BTW do you know how to pseudo-haploidize the data? (preferably in PLINK or EIGENSTRAT format)
Celtion
Regular Member
- Messages
- 77
- Reaction score
- 60
- Points
- 18
No probs – I just found out in the meantime that the majority of ancient samples in the AADR data set have already been pseudo-haplodized. I even double checked by running a .freqx report on a subset of ancient samples in PLINK and they are all homozygous calls.No, sorry.
PaleoRevenge
Well-known member
- Messages
- 1,528
- Reaction score
- 1,344
- Points
- 113
Jovialis
Advisor
- Messages
- 9,901
- Reaction score
- 6,809
- Points
- 113
- Ethnic group
- Italian
- Y-DNA haplogroup
- R1b-PF7566>Y227216
- mtDNA haplogroup
- H6a1b7
Yesssss! Thanks for that
Jovialis
Advisor
- Messages
- 9,901
- Reaction score
- 6,809
- Points
- 113
- Ethnic group
- Italian
- Y-DNA haplogroup
- R1b-PF7566>Y227216
- mtDNA haplogroup
- H6a1b7
Just in case anyone wants to know, you convert eigenstrat format files to plink by creating a parfile in plink like so (This is also a note for me so I can remember; re-learning this stuff after a year can take a while):
1. Open Ubantu and navigate to where you have it mount, and where you send your files. Ming is on a D drive called "UbuntuJovialisHome":
2. Create the parfile, I use nano:
3. In the nano file editor type this:
4. Finally, execute the parfile to produce the BED, BIM, and FAM:
After a while it will finish processing and generate in the destination you are sending it to.
Optional. Here's a quick way to verify in Ubuntu when it is done processing (or you can just navigate there):
1. Open Ubantu and navigate to where you have it mount, and where you send your files. Ming is on a D drive called "UbuntuJovialisHome":
Code:
cd /mnt/d/UbuntuJovialisHome/2. Create the parfile, I use nano:
Code:
nano parfile3. In the nano file editor type this:
Code:
genotypename: /mnt/d/UbuntuJovialisHome/v62.0_HO_public.geno
snpname: /mnt/d/UbuntuJovialisHome/v62.0_HO_public.snp
indivname: /mnt/d/UbuntuJovialisHome/v62.0_HO_public.ind
outputformat: PACKEDPED
genotypeoutname: /mnt/d/UbuntuJovialisHome/data.bed
snpoutname: /mnt/d/UbuntuJovialisHome/data.bim
indivoutname: /mnt/d/UbuntuJovialisHome/data.fam
familynames: NO4. Finally, execute the parfile to produce the BED, BIM, and FAM:
Code:
convertf -p /mnt/d/UbuntuJovialisHome/parfileAfter a while it will finish processing and generate in the destination you are sending it to.
Optional. Here's a quick way to verify in Ubuntu when it is done processing (or you can just navigate there):
Code:
ls -lh /mnt/d/UbuntuJovialisHome/v62.0_HO_public.*